Truhlar Research Group News
June 30, 2026
In a letter that went online at JCTC today:
D. Zhang, Y. Shu, and D. G. Truhlar, Reinventing Density Functional Theory with Machine-Learned Integral Features, doi.org/10.1021/acs.jctc.6c00999
we introduce a conceptually new strategy, going beyond Kohn–Sham theory, in which a multilayer perceptron learns a density functional as a nonlinear function of integral descriptors (“integral features”). In contrast to grid-wise neural network approaches, the integral-features formulation applies the nonlinear model only once per calculation, preserving the computational efficiency of standard density functionals while enabling a substantially richer functional dependence. Using this framework, we develop an integral-features functional, ML25@MN15, trained on databases spanning thermochemistry, kinetics, noncovalent interactions, spin-flip energies, and molecular structures. Enabled by its expanded representational flexibility, the new functional achieves a mean unsigned error of 1.05 kcal/mol over 7232 energetic data from 185 databases and consistently outperforms leading modern functionals across all major categories, including systems containing transition metals. Because the neural network operates on integrated descriptors rather than grid-point values, the computational cost of ML25@MN15 remains comparable to the modern Kohn–Sham functional MN15. These results demonstrate that introducing integral features and allowing the machine to determine the density functional’s dependence on them yields a high-accuracy density functional without increasing computational cost.
Instructions and code for performing the ML25 calculations are provided in the Supporting Information (SI) of the paper. (Link to SI text, Link to code in SI, Link to modified LibXC)
Final author version, click here.
April 15, 2026
Some of group alumni and Jing Xie (alum of the Gagliardi group) got together at the Chinese Chemical Society Congress in Chongqing on April 13th, 2026. Left to right in the standing photo: Linyao Zhang, Ke Yang, Chen Zhou, Xin-Ping Wu, Xuefei Xu, Jing Xie, and Jie Bao. In the seated photo: Ke, Chen, Linyao, Jie, Jing, Xuefei, Xin-Ping. The current Truhlar group sends best wishes.


December 3, 2025
On December 3, Aiswarya M. Parameswaran successfully defended her thesis entitled. “Bridging Accuracy and Efficiency in Multireference Calculations”. The picture shows her post-exam with her defense committee, left to right: Matt Neurock, Don Truhlar, Aiswarya, Kade Head-Marsden, and Doreen Leopold. Best wishes to Aiswarya for her continued success.
One key result in her thesis, from her final project, was showing the success of multiconfiguration pair-density functional theory for the antiaromatic square transition state of cyclobutadiene. With an active space of 8 electrons in 8 orbitals, the six MC-PDFT functionals she tested have an average unsigned error of only 0.8 kcal/mol, with the lowest error (0.2 kcal/mol) being obtained with the MC23 on-top functional. The CASPT2 error is 1.2 kcal/mol with this active space. The 22 Kohn–Sham DFT functionals she tested have an average unsigned error of 6.4 kcal/mol, with the lowest error (2.2 kcal/mol) being obtained with the M06-2X exchange–correlation functional.

December 18, 2024
Suman Bhaumik defended his Ph. D. thesis on December 18, 2024 with a presentation entitled “Application of Density Functional Theory to Heterogeneous Catalysis and Development of Computational Methods for Photochemistry.” Suman is in transition to the role of a Research Scientist at Tokyo Electron Limited US, Albany, NY.

August 19, 2024
The American Chemical Society Division of Computers in Chemistry held a Symposium in honor of Don Truhlar’s birthday at the ACS National Meeting in Denver on August 18 and 19. Don’s lecture was entitled “Improved nonclassical-energy functionals for multiconfiguration nonclassical-energy functional theory (MC-NEFT).” Many group alumni and collaborators presented their research: Yihan Shao, Rubén Meana-Pañeda, Jiali Gao, Hai Lin, Shikha Nangia, Laura Gagliardi, Jingjing Zheng, Arindam Chakraborty, Andreas Heyden, David Schwenke, Hua Guo, Sijia Dong, Roland Lindh, Yan Zhao, Xiaosong Li, Jingzhi Pu, Ahren Jasper, Lucas Bao, Jingyun Ye, Ilja Siepmann, and Ben Janesko.

Don Truhlar with the symposium organizers. Left to right: Hai Lin (University of Colorado at Denver), Don, Sijia Dong (Northeastern University), and Jingzhi Pu (Indiana University at Indianapolis).
August 21, 2023

Whenever possible one prefers to perform dynamical calculations by direct dynamics, in which potential energies, their derivatives, and potential surface couplings are computed on demand by electronic structure calculations carried out side-by-side with the dynamics algorithm. For electronically nonadiabatic processes, this requires not only energies but also nonadiabatic coupling vectors (NACs), which are defined as
$$\mathbf{d}_{I J}=\left\langle\phi_I\middle|\partial / \partial \mathbf{R}\middle| \phi_J\right\rangle$$
where \(\left\langle\ldots\middle|\ldots\middle|\ldots\right\rangle\) denotes an electronic matrix element, \(\phi_I\) and \(\phi_J\) are electronic eigenfunctions, and the operator is a \(3N_\textrm{atoms}\)-dimensional gradient with respect to nuclear coordinates. The evaluation of the NACs adds to the cost, and it is often inconvenient. For example, some software will produce the electronic energies but not the NACs, and for some methods of predicting electronic energies, the NACs are unavailable because the method predicts energies without an explicit wave function corresponding to the energies. In addition to expense and inconvenience, even when NACs are available, they present problems due to origin dependence, spurious long-range couplings, and artificial translational and rotational components.
In some semiclassical multi-surface trajectory methods, like trajectory surface hopping (TSH), one only needs the component of the NAC along the current velocity direction, and in such cases one can use the time derivative of the electronic wave function along the trajectory instead of the NAC. Although, when possible, this is more efficient, it would be even more convenient to have approximations to NACs that can be evaluated straightforwardly from energies and energetic derivatives without gradients of electronic wave functions or time derivatives of electronic wave functions. In other more accurate dynamics methods, like quantum dynamics or coherent switching with decay of mixing (CSDM), one needs the whole NAC. We therefore asked, can we carry out electronically nonadiabatic dynamics using only energy information, and does this provide enough accuracy for efficient propagation of multi-surface molecular dynamics? We were able to answer this question, “YES – NACs for free!”. This work is now published:
“Nonadiabatic Dynamics Algorithms with Only Potential Energies and Gradients: Curvature-Driven Coherent Switching with Decay of Mixing and Curvature-Driven Trajectory Surface Hopping,” Y. Shu, L. Zhang, X. Chen, S. Sun, Y. Huang, and D. G. Truhlar, Journal of Chemical Theory and Computation 18, 1320-1328 (2022). doi.org/10.1021/acs.jctc.1c01080
In the above work, starting with the curvature-based approximation of Baeck and An for one-dimensional systems, we proposed a curvature-approximated time-derivative coupling formula and effective NAC applicable to polyatomic dynamics. (In independent work carried out at about the same time, do Casal et al. [doi.org/10.12688/openreseurope.13624.1] also extended the Baeck–An approximation to multiple dimensions, but in their case only for the component of the NAC along the velocity vector.) We showed how to use the time-derivative coupling formula and effective NACs to obtain curvature-driven versions of CSDM and TSH with energy-based decoherence (TSH-EDC). These curvature-driven methods are respectively called κCSDM and κTSH-EDC. In the original paper (reference above) we compared S1 population decays calculated by κCSDM to those calculated by CSDM based on NACs and CSDM based on time-derivative coupling (approximated by overlap integrals of electronic wave functions). These calculations employed SA-CASSCF electronic wave functions, and the half-lives of the three dynamical methods agree within the statistical uncertainty due to Monte Carlo sampling over initial conditions of the ensemble.
A similar comparison of SA-CASSCF S1 population decays was made for ammonia:
“Direct Nonadiabatic Dynamics of Ammonia with Curvature-Driven Coherent Switching with Decay of Mixing and with Fewest Switches with Time Uncertainty: An Illustration of Population Leaking in Trajectory Surface Hopping Due to Frustrated Hops,” X. Zhao, Y. Shu, L. Zhang, X. Xu, and D. G. Truhlar, Journal of Chemical Theory and Computation 23, 1672-1685 (2023). doi.org/10.1021/acs.jctc.2c01260
Good agreement was found between κCSDM and CSDM based on NACs.
Another check of κCSDM was carried out for photodissociation of 1,3-cyclohexadiene:
“Nonadiabatic Dynamics of 1,3-Cyclohexadiene by Curvature-Driven Coherent Switching with Decay of Mixing,” L. Zhang, Y. Shu, S. Bhaumik, X. Chen, S. Sun, Y. Huang, and D. G. Truhlar, Journal of Chemical Theory and Computation 18, 7073-7081 (2022). doi.org/10.1021/acs.jctc.2c00801
In this work we compared 42-dimensional 3-state κCSDM calculations based on XMS-CASPT2 wave functions to experiment and to earlier dynamics calculations employing NACs [Polyak et al., doi.org/10.1021/acs.jctc.9b00396], and again good agreement was obtained.
Systems with dense manifolds of electronically excited states can be challenging for electronically nonadiabatic dynamics. We compared κCSDM calculations to CSDM calculations based on NACs for total electronically nonadiabatic cross sections in high-energy 5A´collisions of ground-electronic state O2 with ground state O:
Semiclassical Multi-State Dynamics for Six Coupled 5A′ States of O + O2,” F. B. Akher, Y. Shu, Z. Varga, and D. G. Truhlar, Journal of Chemical Theory and Computation 19, 4389-4401 (2023). doi.org/10.1021/acs.jctc.3c00517
These calculations, unlike the ones already discussed, were not direct dynamics but rather were based on analytic fits to diabatic potential energy matrices based on input from 6-state XMS-CASPT2 calculations. We found very good agreement for 30 sets of initial translational and vibrational energies.
Concluding remarks
The curvature-driven methods do not require the calculations of NACs or time derivatives of the wave function. There are no spurious long-range couplings, no origin dependence, no artificial breaking of the conservation of center of mass motion and nuclear angular momentum, no phase issues, and no added expense. Electronically nonadiabatic dynamics calculations agree well with calculations based on NACs. The curvature-driven methods can be used for direct dynamics calculations of electronically nonadiabatic processes with electronic structure methods for which NACs or time derivatives are not available. They simply require adiabatic potential energy surfaces and their gradients. Both κCSDM and κTSH-EDC are available in the software packages SHARC 3.0. and SHARC-MN 2.0:
SHARC 3.0: M. Oppel, maisebastian, and schnarc. (2023). sharc-md/sharc: SHARC Release 3.0.1 (v3.0.1). Zenodo. https://doi.org/10.5281/zenodo.7896064
SHARC-MN 2.0: Y. Shu, L. Zhang, and D. G. Truhlar. (2023). SHARC-MN v2.0 (2.0). Zenodo. https://doi.org/10.5281/zenodo.7818894

August 11, 2023
The editors of Chemical Physics Reviews evaluated the article “Computational Quantum Chemistry of Metal–Organic Frameworks” by Indrani Choudhuri, Jingyun Ye, and Donald G. Truhlar [Chem. Phys. Rev. 4, article no. 031304 (2023); doi.org/10.1063/5.0153656] as one of the journal’s best, and they chose to promote it as a Feature Article. The article is discussed in “Computational methodologies hunt for elusive metal-organic frameworks” by Amy Thompson [Scilight 2023 (32); doi.org/10.1063/10.0020716].
Metal–organic frameworks (MOFs) exhibit exceptional properties suitable for a wide range of applications, including gas separation and storage, heterogeneous catalysis, photocatalysis, and electrocatalysis. Computational modeling has emerged as a valuable and efficient tool for comprehending and designing MOFs and explicating their functional behavior. In this review, we outline various computational methodologies, especially Kohn–Sham density functional theory, for determining the structural, electronic, and magnetic properties of MOFs, and we survey selected illustrative applications of these methods. The combination of state-of-the-art computational methods reviewed in the article with more comprehensive experimental data obtained through advanced instrumentation and characterization techniques is expected to yield a deeper understanding of current MOF applications and enable rational design of MOFs with enhanced functionalities.

June 21, 2023
Several former group members and friends of the group attended the 33rd Chinese Chemical Society Congress in Qingdao this month. The attached photo shows (left to right):
Ke Yang, Naikan University
Xin Zhang, Beijing University of Chemical Technology
Xuefei Xu, Tsinghua University
Peifeng Su, Xiamen University
Xiao He, East China Normal University
Bo Long, Guizhou Minzu University
Wei Lin, Fuzhou University
Xin-Ping Wu, Shanghai Jiao Tong University
Jinfeng Liu, China Pharmaceutical University (previous PhD student of Xiao He)
Our group is very proud of the continued success of all our group alumni and friends.

May 24, 2023
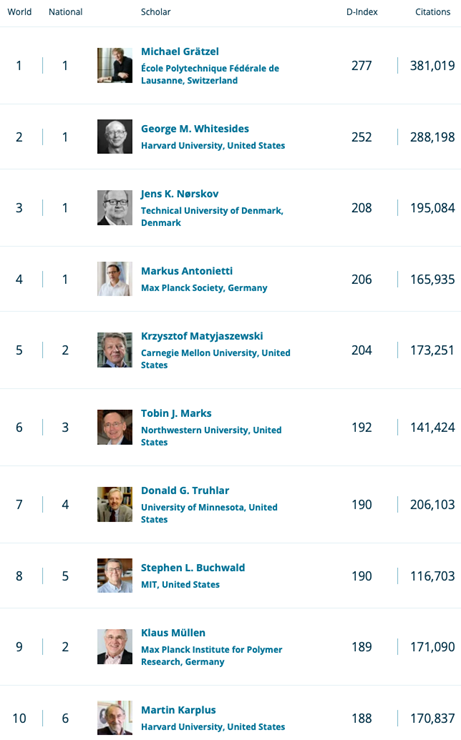
Yesterday, Research.com informed Professor Truhlar that he has been recognized with their Chemistry Leader Award for 2023. Research.com recently released the 2023 Edition of the Ranking of Top Scientists in the field of Chemistry:
https://research.com/scientists-rankings/chemistry
Professor Truhlar ranked 7th in the world and 4th in United States.
January 9, 2023
Today was published our invited perspective article in JCTC:
Y. Shu and D. G. Truhlar, Journal of Chemical Theory and Computation 19, 380-395 (2022).
It is openly available to all readers at doi.org/10.1021/acs.jctc.2c00988.
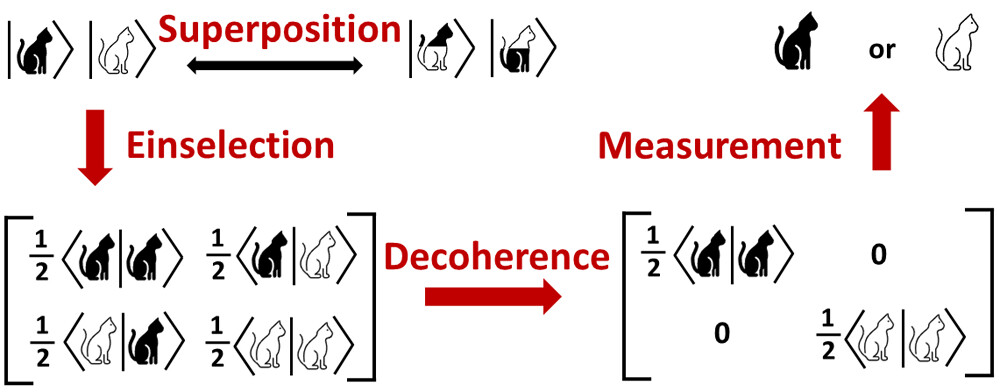
In this perspective, we introduced the theoretical concepts that are essential to understand decoherence in electronically nonadiabatic molecular events. We emphasized that the reduced density matrix of a subsystem evolves according to a nonunitary Liouville–von Neumann equation even if the full density matrix of the combined subsystem and environment remains pure. When governed by a nonunitary Liouville–von Neumann equation, the off-diagonal elements of the subsystem reduced density matrix decay to zero. This is the central fact of decoherence; and this decay of coherence is essential in understanding the propagation of the electronic reduced density matrix in chemical and physical systems, not just in a condensed phase due to the solvent but even in a small molecule in the gas phase, where the electronic subsystem is decohered by the nuclei, which act as an environment. We discussed how the continuous monitoring of electrons by the nuclei causes decoherence of the reduced electronic density matrix.
Simulations of the decoherence effect in nonadiabatic dynamics can be achieved by a combination of a decay-of-mixing algorithm and an ensemble average over initial conditions, for example by decoherence with decay of mixing, which is available in the ANT program and will soon become available in SHARC-3.0.
Decoherence was also recently discussed (very briefly) in Don Truhlar’s letter to the editor of Physics Today: “More on the Quantum Measurement Problem,” D. G. Truhlar, Physics Today 75(11), 13 (Nov. 2022) doi.org/10.1063/PT.3.5113