
Truhlar Research Group News
August 7, 2019
In June, our group was pleased to welcome Lyuben Borislavov from the University of Sophia in Sophia, Bulgaria, where he was recommended by Professors Alia Tadjer and Todor Dudev, and Erica Mitchell from Taylor University in Upland Indiana, where she was recommended by Professor T. Brandon Magers. Erica worked on the multi-configuration pair-density functional theory of Diels-Alder reactions and the speedup of MC-PDFT analytic gradients, co-supervised by Professor Laura Gagliardi and also aided by Thais Scott, Jie Bao, and Matt Hermes. Lyuben worked on machine learning of molecular dynamics trajectories for the photodissociation of thioanisole, aided by Kelsey Parker and Yinan Shu. Both students presented their work at the 2019 Summer Undergraduate Research Fellowship Symposium (SURF Symposium) of CTC on August 6: The speakers were
Lyuben Borislavov from Sofia University (Bulgaria), Don Truhlar group
Janey Lin from Mount Holyoke College, Massachusetts, Laura Gagliardi group
Maria Mikhailenko from ITMO University (Russia), Jason Goodpaster group
Erica Mitchell from Taylor University, Indiana, groups of Gagliardi and Truhlar
Claire Shugart from Carlton College, Minnesota, Gagliardi group
Samuel Powell from Ohio Northern University, Gagliardi group
Victoria Vernadskaia from ITMO University (Russia), Ilja Siepmann group
Peiyao Wu from Emory University, Georgia, Siepmann group
After the symposium the picture at the right was taken of all the summer researchers in the CTC SURF program, along with the participating mentors and advisers.
July 31, 2019
The Royal Society of Chemistry has informed us that three papers from our group are all in the top 5% of highly cited papers in their General chemistry portfolio of journals. The successful articles are as follows:
|
|
Article |
Times cited in 2018 |
|
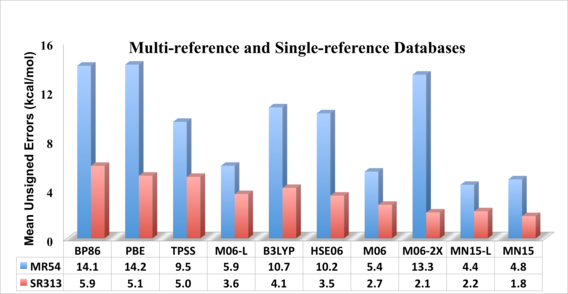 doi.org/10.1039/C6SC00705h |
“MN15: A Kohn-Sham Global-Hybrid Exchange-Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions,” H. S. Yu, X. He, S. L. Li, and D. G. Truhlar, Chemical Science 7, 5032-5051 (2016).
|
59 |
|
 doi.org/10.1039/C7CS00602K |
“Variational Transition State Theory: Theoretical Framework and Recent Developments,” J. L. Bao and D. G. Truhlar, Chemical Society Reviews 46, 7548-7596 (2017).
|
23 |
|
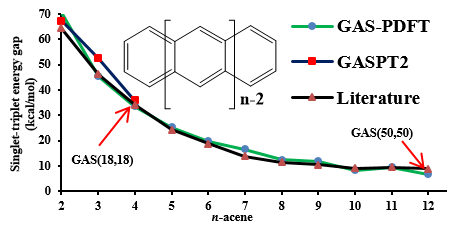 doi.org/10.1039/C6SC05036K |
“Generalized-Active-Space Pair-Density Functional Theory: An Efficient Method to Study Large, Strongly Correlated, Conjugated Systems,” S. Ghosh, C. J. Cramer, D. G. Truhlar, L. Gagliardi, Chemical Science 8, 2741-2750 (2017).
|
14 |
February 17, 2019

Postdoctoral associate Jingyun Ye, a joint member of the Truhlar and Cramer groups, will begin as assistant professor of Chemistry at Clarkson University in Potsdam, New York in January 2020, and we wish her the best in her new opportunity. Meanwhile we look forward to ten months of further work on projects in the Inorganometallic Catalyst Design Center.
February 6, 2019
The editors of PCCP (the physical chemistry journal of the Royal Society of Chemistry) have selected the paper
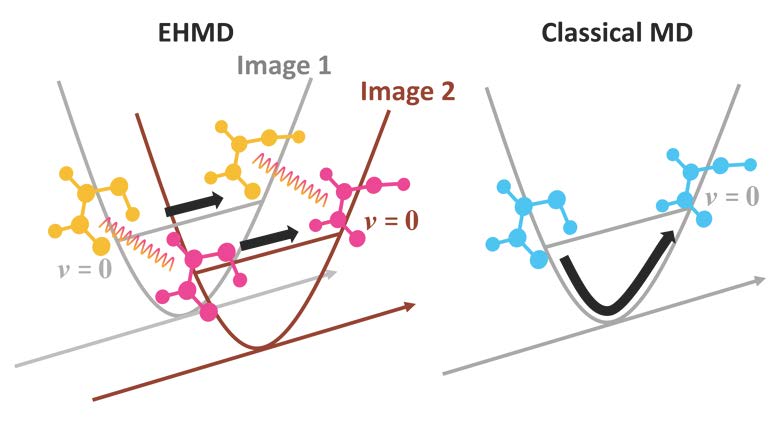
“Extended Hamiltonian Molecular Dynamics: Semiclassical Trajectories with Improved Maintenance of Zero Point Energy,” Yinan Shu, Sijia S. Dong, Kelsey A. Parker, J. Lucas Bao, Linzao Zhang, and Don Truhlar, Physical Chemistry Chemical Physics 20, 30209-30218 (2018). doi.org/10.1039/C8CP04914A
for the the themed collection "2018 PCCP HOT Articles”.
Classical molecular dynamics is one of the most widely employed simulation tools in physical chemistry. A long-standing problem in classical molecular dynamics simulations is the inability of classical mechanics to maintain zero point energy in the vibrational modes. There have been many attempts to overcome this deficiency, but no practical and completely satisfactory method has emerged. This article shows how the problem can be tamed by presenting a very simple extended Hamiltonian goes a long way toward solving the problem. The paper is of broad interest because the method can be applied even to the most complex problems for which classical simulations are currently employed.
August 10, 2018

Junwei (Lucas) Bao successfully defended his Ph D. thesis on August 10, 2018. His thesis is entitled Reaction Rate Theory, Electronic Structure Theory, and Applications; it has an introduction plus 19 chapters covering his publications. Congratulations Lucas! Lucas will now head out to Princeton for a postdoctoral position with Professor Emily Carter.
The picture shows his committee immediately after the defense, from left to right: Tom Schwartzentruber, Don Truhlar, Lucas Bao, Aaron Massari, and Chris Cramer.
June 2, 2018
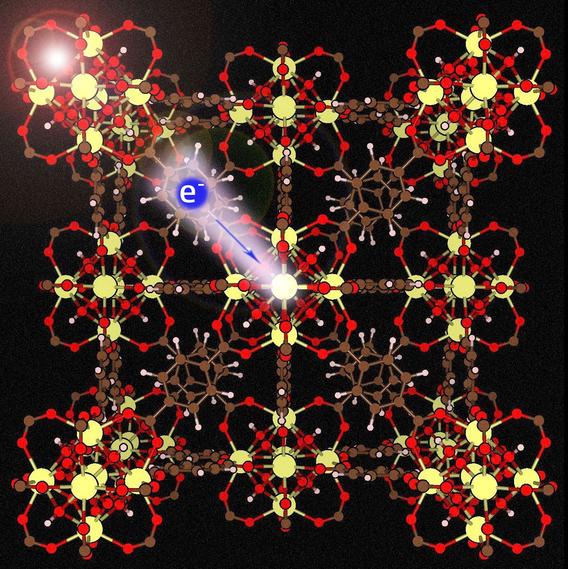
Photocatalysts can absorb light and convert the exciton into an electron–hole pair, so the electron can drive a reduction reaction and/or the hole can drive an oxidation reaction. Photocatalysts are usually inorganic semiconductors (e.g., TiO2). Dr. Xin-Ping Wu, a postdoctoral scholar working with Don Truhlar and Laura Gagliardi, has proposed that metal–organic frameworks (MOFs) containing cerium would also be good photocatalysts. MOFs are crystalline materials with some attractive properties for photocatalysis, including well separated inorganometallic nodes that can serve as catalytic centers and – since MOFs are nanoporous –including high accessibility of potential catalytic sites to reagents; however, the highest unoccupied and the lowest unoccupied crystal orbitals (HOCO and LUCO) of MOFs are usually localized solely on their linkers, so most MOFs have poor charge separation capabilities, which leads to short lifetimes of excited states and limits possible photocatalytic activity. But Ce has low-lying empty 4f orbitals, and Ce-MOFs can have the LUCO on Ce in the node, thereby separating the HOCO and LUCO onto different subsystems (the linker and the node) in the MOF, as shown in the figure on the right. This anticipated result was confirmed by quantum chemical calculations on a substituted UiO-66 MOF, with Zr (for stability) and Ce (for photocatalytic activity) as the metals in the nodes. With purposefully designed linkers, absorption is in the visible and the absolute orbital energies are well situated for water splitting or CO2 reduction. The work shows the power of theory in engineering the electronic properties of functional nanoporous materials.
The work is published in the Journal of the American Chemical Society [doi.org/10.1021/jacs.8b03613]. The research was supported by the Nanoporous Materials Genome Center (NMGC) [NMGC main page], which is a Computational Chemical Sciences Center in the Department of Chemistry at the University of Minnesota.
February 20, 2018

Jingyun Ye received an award for her poster, “Computational Study of MOF-Supported Metal Catalysts for Ethylene Dimerization,” at the conference on New Challenges in Heterogeneous Catalysis at the King Abdullah University of Science and Technology (KAUST) in Thuwal, Saudi Arabia, Monday, Jan. 29. The poster reported computational simulations to design Ni catalysts supported on the NU-1000 metal organic framework and to identify the catalytically active sites for the ethylene dimerization reaction. Furthermore, she reported screening of a variety of metals to identify potential catalyst candidates for ethylene dimerization in order to increase the selectivity for 1-butene production.
December 16, 2017

Junwei Lucas Bao has been named as one of two recipients of the 2018 Graduate Award in Theoretical Chemistry given by the American Chemical Society Physical Chemistry Division. Lucas is honored for his work in several areas, especially chemical kinetics and transition-metal bond dissociation energies. He will present an award lecture at the Spring National ACS Meeting in New Orleans. Congratulations Lucas!
September 15, 2017

Congratulations to Chad Hoyer who successfully defend his thesis today. Chad was co-advised by Professors Laura Gagliardi and Don Truhlar. His thesis is entitled "Electronic Structure Method Development for Excited-State Chemistry” and it includes research on multi-configuration pair-density functional theory for excited states and on dipole-quadrupole-electrostatic potential (DQΦ) diabatization. The picture at the right shows Laura Gagliardi, Chad Hoyer, and Don Truhlar after Chad’s defense.