Truhlar Research Group News
May 26, 2020
The languages that physicists and chemists use in regarding the HOMO-LUMO gaps, HOCO-LUCO gaps, band gaps, fundamental gaps, and optical excitation thresholds can be quite different, and the difference in language has the very serious consequence that it changes the way that different communities interpret their calculations. We studied the relations between these kinds of quantities for a large number of exchange-correlation functionals. Surprisingly, despite large differences in functional forms, a consistent relation with the percentage of Hartree-Fock exchange was discovered. Furthermore, we concluded that with presently available functionals the orbital energies should be treated as intermediate mathematical variables in the calculation of excitation energies rather than as the energies of independent-particle reference states for quasiparticle theory.
“Relationships between Orbital Energies, Optical AND fundamental Gaps, Exciton Shifts in Approximate Density Functional Theory and Quasiparticle Theory” Y. Shu, and D. G. Truhlar, Journal of Chemical Theory and Computation 16, 7 (2020). doi,org/10.1021/acs.jctc.0c00320
May 7, 2020
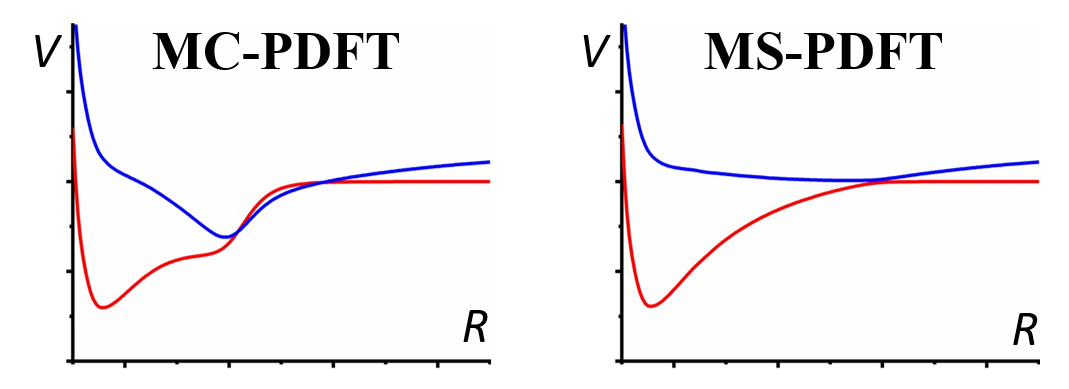
When two electronic states of the same symmetry are close in energy, they have strong interactions, and they cannot be treated separately – one must use multi-state methods. To incorporate state interactions in multiconfiguration pair-density functional theory (MC-PDFT), we have developed two new multi-state (MS) methods, namely extended multi-state PDFT (XMS-PDFT) and variational multi-state PDFT (VMS-PDFT. The new methods have been tested successfully for eight systems exhibiting locally avoided crossings among two to six states. Since both XMS-PDFT and VMS-PDFT are much less expensive than XMS-CASPT2, they will allow well-correlated calculations on large systems for which perturbation theory is undoable. The figure shows schematically how nonphysical results are obtained for potential curves when they are treated separately – on the left, and how this is remedied when they are treated together by a multi-state method – on the right.
“Multi-state pair-density functional theory,” J. J. Bao, C. Zhou, Z. Varga, S. Kanchanakungwankul, L. Gagliardi and D. G. Truhlar, Faraday Discussions 224, 348-372 (2020). doi.org/10.1039/D0FD00037J
April 16, 2020

Yinan Shu, Ph.D., a post-doctoral associate working with Regents Professor Donald Truhlar, has received the American Chemical Society Division of Physical Chemistry's 2020 Young Investigator Award. He was one of four researchers selected to receive this award and will present his research at a future PHYS symposium.
Yinan earned his bachelor's degrees in chemistry and biological science from Wuhan University in China, and his doctorate from Michigan State University. His thesis focused on, "Understanding Non-Radiative Recombination Processes of the Optoelectronic Materials from First Principles." His research work with Professor Truhlar focuses on understanding complex behaviors of molecules and materials upon excitations and employ machine learning to solve theoretical chemistry problems.
This news article was written by Eileen Harvala for the Department of Chemistry News.
April 6, 2020
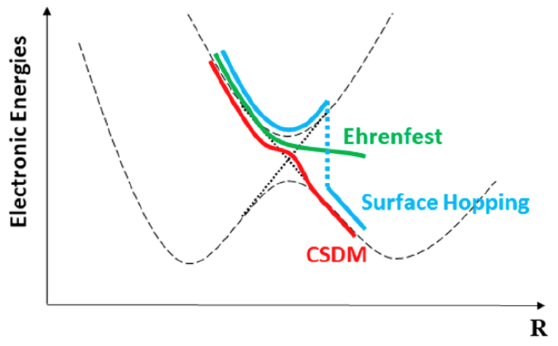
In collaboration with Leticia González and Sebastian Mai, we have implemented coherent switching with decay of mixing (CSDM) into the SHARC program. CSDM is a semiclassical nonadiabatic dynamics algorithm that incorporates advantages from both surface hopping and the semiclassical Ehrenfest method, and it allows a more accurate treatment than other semiclassical methods of the competition between coherence and decoherence. One can obtain our current modified SHARC program from our group software page: https://comp.chem.umn.edu/sharc-mn/. Now CSDM is can be used to study both internal conversion and intersystem crossing processes by using the SHARC approach, and it is easier to interface with electronic structure programs for direct dynamics.
“Implementation of Coherent Switching with Decay of Mixing into the SHARC Program” Y. Shu, L. Zhang, Z. S. Mai, S. Sun, L. González and D. G. Truhlar, Journal of Chemical Theory and Computation 16, 3464 (2020). doi.org/10.1021/acs.jctc.0c00112
March 24, 2020
We are pleased to congratulate two group alumni on new positions that they will assume starting in academic year 2020-2021. Junwei Lucas Bao, who was a graduate student in our group from 2014 to 2018 and who is now a postdoctoral associate with Emily Carter at Princeton, has accepted a position as assistant professor in the Department of Chemistry at Boston College in Chestnut Hill, MA. Dr. Sijia Dong, who was a postdoctoral associate in our group from 2017 to 2019 and who is now a postdoctoral associate in the group of Guilia Galli at the University of Chicago, has accepted an assistant professor position in the Department of Chemistry and Chemical Biology at Northeastern University in Boston, MA. We wish them both well, and we are confident they will excel in their new positions!


March 2, 2020
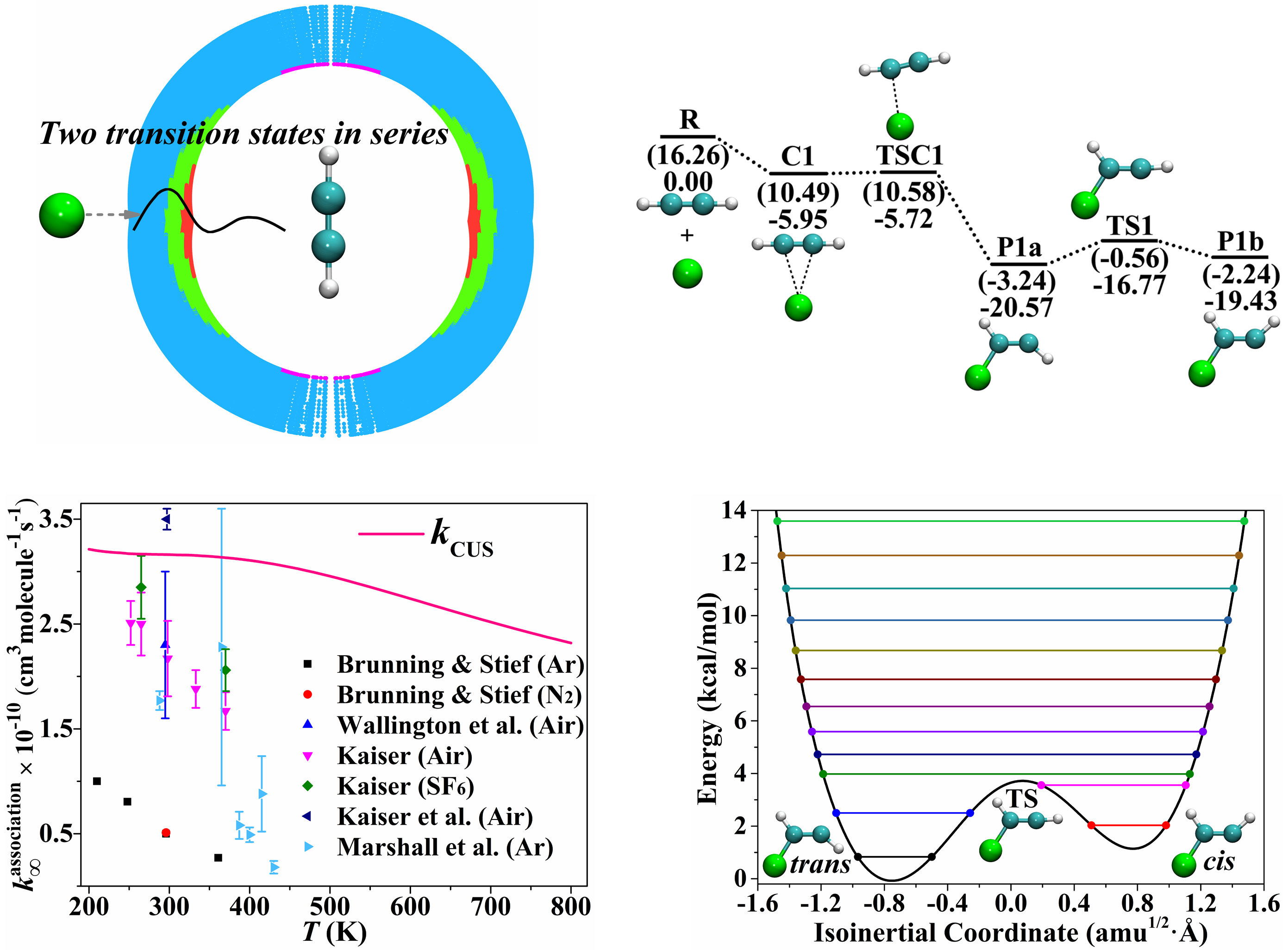
A key issue in chemical kinetics is advancing the theoretical framework to handle reactions beyond the domain of textbook transition state theory by including – for example – anharmonicity, barrierless transition states, transition states in series, and the effect of conformational flexibility on equilibrium constants. Other key issues are validating affordable electronic structure methods for direct dynamics and the direct calculation of high-pressure limiting rate constants, which are often obtainable experimentally only by extrapolation. We have addressed all these issues in a study of the prototype radical–molecule barrierless association reaction Cl + C2H2, and this work demonstrates the ability of recent advances in theoretical methods, when combined, to provide rate constants even for a difficult class of reactions in cases where experimental data are uncertain or missing.
“Association of Cl with C2H2 by Unified Variable-Reaction-Coordinate and Reaction-Path Variational Transition State Theory,” L. Zhang, D. G. Truhlar, and S. Sun, Proceedings of the National Academy of Sciences U.S.A. 117, 5610-5616 (2020). doi.org/10.1073/pnas.1920018117
February 6, 2020
The Journal of Chemical theory and Computation has published a virtual issue honoring the work of the Truhlar group and collaborators:
https://pubs.acs.org/page/jctcce/vi/truhlar.html
The coauthors honored in this virtual issue are Ahren Jasper, Alek Marenich, Andy Luo, Antonio Fernández-Ramos, Becky Carlson, Ben Janesko, Bo Wang, Chao Zhu, Chris Cramer, Dongxia Ma, Erin (Dahlke) Speetzen, Ewa Papajak, Giovanni Li Manni, Giovanni Scalmani, Hannah Leverentz, Haoyu Yu, Jeppe Olsen, Jingjing Zheng, John Alecu, Kaining Duanmu, Ke Yang, Laura Gagliardi, Lucas Bao, Mike Frisch, Pragya Verma, Rubén Meana-Pañeda, Samuel Odoh, Shaohong L. Li, Steve Jerome, Xiao He, Xuefei Xu, Yan Zhao, and Ying Wang.
January 20, 2020
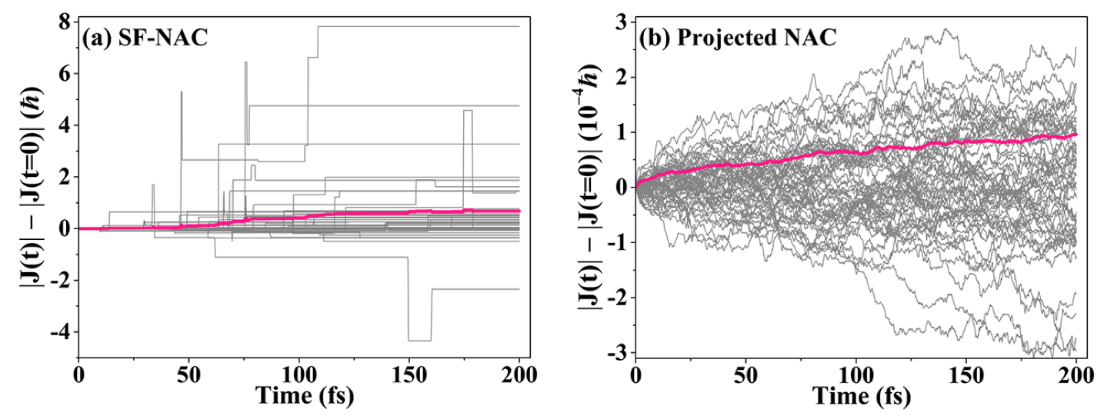
We found that commonly used semiclassical nonadiabatic dynamics, namely trajectory surface hopping, semiclassical Ehrenfest, and coherent switching with decay of mixing (CSDM) fail to conserve nuclear angular momenta in direct dynamics. The failure is caused by the direct use of the space-frame (SF) for the nonadiabatic coupling vector (NAC) in standard electronic structure software. We showed how to eliminate the problem by using a projected NAC, in which translational and rotational parts are removed. The plots illustrate how projection reduces the problem by four orders of magnitude.
“Conservation of Angular Momentum in Direct Nonadiabatic Dynamics” Y. Shu, L. Zhang, Z. Varga, K. A. Parker, S. Kanchanakungwankul, S. Sun, and D. G. Truhlar, Journal of Physical Chemistry Letters 11, 1135 (2020). doi.org/10.1021/acs.jpclett.9b03749
September 11, 2019

Our group recently welcomed postdoctoral associate Dihua Wu and visiting student Shuhang Li to the group. The montage photo that accompanies this news item shows all the Nankai University members and past members of the group. At the upper left is Professor Ruifang Li of Nankai University, who first entered Nankai in 1999 and who was the first Nankai University participant in the Truhlar group, starting in 2009. At the upper right is Benchen (Kevin) Huang, now a graduate student at the University of Chicago, who entered Nankai in 2015 and was in our group as a visiting student in 2018-2019. In front of the bookcase are current group members, Dihua Wu (first left, entered Nankai in 2008), Jiaxin Ning (second left, entered Nankai in 2014, now a graduate student in the group), Don Truhlar (middle), Shuhang Li (second right, entered Nankai in 2016), and Jie Bao (first right, entered Nankai in 2010, now a graduate student in the group).