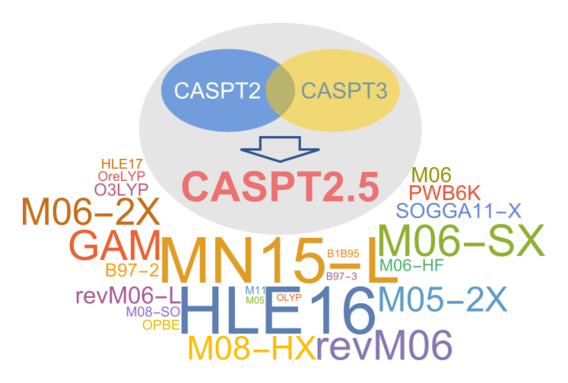
Our recent benchmark study shows that the Minnesota exchange–correlation density functionals MN15-L, M06-SX, and revM06 can predict highly accurate spin splitting energies for transition metals. The def2-TZVP basis set is found to be the most suitable basis set within our test set to perform these density functional calculations. We also proposed a new wave function method called CASPT2.5, which is performed by taking the average of the CASPT2 and CASPT3 energies. We found that CASPT2.5 extrapolated to a complete basis set is the best wave function method in terms of computational cost and accuracy for spin splittings of transition metals.
“Spin Splitting Energy of Transition Metals: A New, More Affordable Wave Function Benchmark Method and Its Use to Test Density Functional Theory,” D. Zhang and D. G. Truhlar, Journal of Chemical Theory and Computation 16, 4416-4428 (2020). doi.org/10.1021/acs.jctc.0c00518